
BSTMatrix v2.3.R环境搭建、安装及使用说明

百创DG1000—某野生动物脑细胞核
2022年5月31日
BSTViewer_v3.0软件安装图文教程
2022年6月8日生成矩阵
1.1 软件环境依赖
A)python:版本8及以上,安装cv2模块(4.0以上版本),matplotlib,seaborn,pandas(1.5.0以上版本),tifffile,Cellpose,Pyyaml,scikit-image,tqdm
B)Star:版本6.1d及以上版本
C)perl:threads及 threads::shared 模块
D)Rscript:需要以下 R 包 ,seurat 、dplyr 、tibble 、ggplot2 、broom 、purrr 、cowplot 、cluster 、ggpubr、 plotly 、htmlwidgets 、kableextra 、htmltools 、shiny、knitr 、rmarkdown、optparse 、getopt。
E)Pandoc。
【依赖的软件需要使用 export 添加到环境变量中, 以实现流程的调用】
【环境部署可参考安装目录下“环境配置.rst”文件】
1.2 输入数据
A)测序数据:双端测序 fastq 数据。
B)参考基因组数据:基因组序列文件 ,gtf 文件(第 3 列需要包含 exon) ,gff 文件(可选)(第 3 列 需要包含 gene、exon)。
C)tsv文件:可使用 gtf 文件生成,参考命令:perl ./tools/features_generate.pl -i xxx.gtf-o features.tsv
D)STAR基因组索引文件: 可使用基因组序列文件和 gtf 文件生成, 参考命令 :STAR –runThreadN 8 –runMode genomeGenerate –genomeDir star/ –genomeFastaFiles genome.fa –sjdbGTFfile gtf
E)荧光解码文件及HE 图片文件
F)荧光图片文件(HE和荧光图片至少给定1个)。
1.3 配置文件( config.txt)
config.txt:
## fq 测序数据文件路径, 支持.gz 格式
FQ1 /path/to/read_1.fq.gz
FQ2 /path/to/read_2.fq.gz
## Flu info file 荧光解码文件路径
FLU /path/to/flu_info.txt
## AllheStat.py 组织识别参数
HE /path/to/HE.tif #HE 染色图片 ,和组织荧光图片至少给一个
#INSIDE 0 #是否对组织内部空白进行识别,0不识别,1识别
#GRAY 200 #组织图像识别灰度阈值,默认自动判断
## CellSplit 是否做细胞分割及荧光图片路径、颜色通道
CellSplit True #是否做细胞分割分析,T/True 表示做 ,否则不做
Fluorescence /path/to/fluorescence.tiff #组织荧光图片,可选。
fluorescence_channl 0 #荧光图片颜色通道,默认0
#FLGRAY 15 #荧光图片识别灰度阈值,默认自动判断
#cells_npy /path/to/cells/npyfile #已有细胞分割pny结果文件,给定则用此文件分析
#YAML /path/to/cell_split/parameter/file #细胞分割参数文件,可选
## 参考基因组 STAR 建库目录及 gff/gtf 文件路径
GenomeVer xxx #基因组版本信息,报告中使用
INDEX /path/to/STAR/index/dir/
GFF /path/to/ref/gene/gff3/file #( 也可使用 gtf 文件)
## 参考基因组 features.tsv 文件路径
FEATURE /path/to/features.tsv
## 输出目录及输出文件前缀
OUTDIR /path/to/result/dir/
PREFIX outfile-prefix
### 程序参数
## fastq2BcUmi
BCType V2 #barcode 版本类型(一般为 V2 版本)
BCThreads 8 #线程数
## Umi2Gene
Sjdboverhang 100 #STAR 建库时使用的-sjdboverhang 参数值 ,默认 100
STARThreads 8 #STAR 比对线程数
## ENV python 和 Rscript 的路径 ,如不提供则使用系统环境中的版本(不提供请注释掉以下参数)
PYTHON /path/to/python/dir/
Rscript /path/to/Rscript/dir/
注:要进行细胞分割分析配置文件中CellSplit需选True,植物细胞分割不需要提供荧光图,只需在HE参数中给定明场图,并且注释掉荧光图参数Fluorescence 。
1.4 流程说明
1.4.1 流程分为 8 个步骤 ,如下所示:
步骤 1:运行 fastq2BcUmi ,识别 fastq 数据中的 barcode 、umi。
步骤 2:运行 LinkBcChip ,识别荧光数据的 barcode 信息并对应到芯片上位置。
步骤 3:运行 Umi2Gene ,将 reads 与参考基因组比对 ,得到每个 UMI 对应的基因信息。 步骤 4:运行 MatrixMake ,获得基因表达矩阵。
步骤 5:运行 AllheStat, 处理 HE 图片。
步骤 6:运行 cluster.R ,进行聚类分析。
步骤 7:运行CellSplit,进行细胞分割分析。
步骤 8:运行 WebReport ,得到网页版报告。
1.4.2 流程参数:
-c config.txt 数据配置文件
-s 步骤选择,0 为运行 1-8 所有步骤,也可选择个别步骤单独运行 ,多个步骤中间使用“,”分隔 。
注: 选 0 时如果要做细胞分割分析, 配置文件中 CellSplit 参数需要选 True。
注:植物细胞分割时不需要荧光图,只需要在配置文件的HE参数中提供明场图,CellSplit参数选True即可进行分析。
1.4.3 参考命令:
./BSTMatrix -c config.txt -s 0
./BSTMatrix -c config.txt -s 1,2,3,4,5,6,7,8
./BSTMatrix -c config.txt -s 1,3
1.5 结果文件说明
目录结构及结果说明:
outdir/
├── 01.fastq2BcUmi 步骤1 序列barcode及umi检测结果目录
│ ├── xxx.bc_dist 不同barcode检测统计文件
│ ├── xxx.bc_stat 不同barcode检测统计文件
│ ├── xxx.bc_umi_read.tsv barcode类型、对应的umi及reads数统计文件
│ ├── xxx.bc_umi_read.tsv.id barcode类型、对应的umi及reads id文件
│ ├── xxx.filter 没有完整识别出barcode的reads信息文件
│ ├── xxx.full_stat barcode类型对应的reads数、umi数文件
│ ├── xxx.id_map id编号对应关系文件
│ ├── xxx.qual.stat reads统计文件
│ ├── xxx.select_id 完整识别出barcode和UMI的reads id文件
│ ├── xxx.stat barcode检测统计文件
│ ├── xxx.umi reads对应的barcode类型及umi文件
│ └── xxx.umi_cor.info umi校正信息文件
├── 02.LinkBcChip 步骤2 barcode空间定位结果目录
│ ├── xxx.barcode_pos.tsv barcode类型对应的芯片位置文件
│ ├── xxx.barcode.tsv 芯片对应的barcode类型文件
│ ├── xxx.flu.stat barcode解码统计文件
│ ├── xxx.info 芯片位置对应的barcode信息文件
│ └── xxx.null 无法识别的芯片位置信息文件
├── 03.Umi2Gene 步骤3 基因表达信息结果目录
│ ├── xxxAligned.sortedByCoord.out.bam STAR比对结果bam文件
│ ├── xxx.cut0.fq 选择进行比对的read2序列文件
│ ├── xxxLog.final.out STAR比对统计输出文件
│ ├── xxxLog.out STAR比对日志文件
│ ├── xxxLog.progress.out STAR比对日志文件
│ ├── xxx.map2gene 比对到基因上的reads信息文件
│ ├── xxxSJ.out.tab STAR比对的剪切位点信息文件
│ ├── xxx_STARtmp STAR比对临时文件目录
│ ├── xxx.stat 初步比对信息统计文件
│ ├── xxx.total.stat 比对情况统计文件
│ └── xxx.umi_gene.tsv barcode对应的umi及基因文件
├── 04.MatrixMake 步骤4 表达矩阵结果目录
│ ├── xxx.matrix.tsv 基因表达矩阵文件
│ ├── xxx.matrix.tsv.filt 过滤文件
│ ├── xxx.select.bc_umi_read.tsv barcode对应的umi及reads数文件
│ ├── xxx.select.umi_gene.tsv barcode对应的umi及基因文件
│ ├── xxx.select.umi_gene.tsv.filter 过滤掉的barcode基因信息文件
│ ├── xxx.sequencing_saturation.stat 测序饱和度统计文件
│ └── xxx.sequencing_saturation.png 测序饱和度曲线图
├── 05.AllheStat 步骤5 组织表达分析结果目录
│ ├── allhe 组织区域信息目录
│ │ ├── he_roi_small.png HE组织区域识别后png图片文件
│ │ ├── he_roi.tif HE组织区域识别后tif图片文件
│ │ ├── roi_heAuto.json 组织区域json文件
│ │ └── stat.txt 组织区域统计文件
│ ├── all_level_stat.txt 不同分辨率水平统计文件
│ ├── BSTViewer_project BSTViewer软件输入数据目录
│ │ ├── cell_split 细胞分割数据目录
│ │ ├── cluster 空
│ │ ├── he_roi_small.png HE组织区域识别后png图片文件
│ │ ├── he.tif HE染色图片文件
│ │ ├── imgs 空
│ │ ├── level_matrix 不同分辨率水平表达矩阵目录
│ │ ├── project_setting.json BSTViewer项目json文件
│ │ ├── roi_groups 组织及HE图片json文件目录
│ │ └── subdata 组织区域不同分辨率水平表达矩阵目录
│ ├── heAuto_level_matrix 组织区域不同分辨率水平表达矩阵目录
│ │ └── subdata 组织区域不同分辨率水平表达矩阵目录
│ ├── level_matrix 芯片不同分辨率水平表达矩阵目录
│ │ ├── level_1 level 1水平表达矩阵目录
│ │ ├── level_13 level 13水平表达矩阵目录
│ │ ├── level_2 level 2水平表达矩阵目录
│ │ ├── level_3 level 3水平表达矩阵目录
│ │ ├── level_4 level 4水平表达矩阵目录
│ │ ├── level_5 level 5水平表达矩阵目录
│ │ ├── level_6 level 6水平表达矩阵目录
│ │ └── level_7 level 7水平表达矩阵目录
│ ├── stat.txt 组织区域分析统计文件
│ └── umi_plot umi画图结果目录
│ ├── all_umi_count_small.png 芯片区域umi-count png图片
│ ├── all_umi_count.tif 芯片区域umi-count tif图片
│ ├── roi_umi_count_small.png 组织区域umi-count png图片
│ ├── roi_umi_count.tif 组织区域umi-count tif图片
│ ├── roi_umi_count_white_small.png 组织区域白底umi-count png图片
│ └── roi_umi_count_white.tif 组织区域白底umi-count tif图片
├── 06.Cluster 步骤6 聚类分析结果目录
│ ├── L13 level 13水平聚类结果目录
│ │ ├── cluster.csv 聚类结果文件
│ │ ├── L13_cluster_files 聚类html附录文件目录
│ │ ├── L13_cluster.html 聚类html格式图片文件
│ │ ├── L13_cluster.pdf 聚类pdf格式图片文件
│ │ ├── L13_cluster.png 聚类png格式图片文件
│ │ ├── L13_umap_clstr.pdf 合并后pdf格式图片文件
│ │ ├── L13_umap_clstr.png 合并后png格式图片文件
│ │ ├── L13_umap_files umap html附录文件目录
│ │ ├── L13_umap.html umap html格式图片文件
│ │ ├── L13_umap.pdf umap pdf格式图片文件
│ │ └── L13_umap.png umap png格式图片文件
│ ├── L3 level 3水平聚类结果目录
│ │ ├── cluster.csv 聚类结果文件
│ │ ├── L3_cluster_files 聚类html附录文件目录
│ │ ├── L3_cluster.html 聚类html格式图片文件
│ │ ├── L3_cluster.pdf 聚类pdf格式图片文件
│ │ ├── L3_cluster.png 聚类png格式图片文件
│ │ ├── L3_umap_clstr.pdf 合并后pdf格式图片文件
│ │ ├── L3_umap_clstr.png 合并后png格式图片文件
│ │ ├── L3_umap_files umap html附录文件目录
│ │ ├── L3_umap.html umap html格式图片文件
│ │ ├── L3_umap.pdf umap pdf格式图片文件
│ │ └── L3_umap.png umap png格式图片文件
… …
│ └── L7 level 7水平聚类结果目录
│ ├── cluster.csv 聚类结果文件
│ ├── L7_cluster_files 聚类html附录文件目录
│ ├── L7_cluster.html 聚类html格式图片文件
│ ├── L7_cluster.pdf 聚类pdf格式图片文件
│ ├── L7_cluster.png 聚类png格式图片文件
│ ├── L7_umap_clstr.pdf 合并后pdf格式图片文件
│ ├── L7_umap_clstr.png 合并后png格式图片文件
│ ├── L7_umap_files umap html附录文件目录
│ ├── L7_umap.html umap html格式图片文件
│ ├── L7_umap.pdf umap pdf格式图片文件
│ └── L7_umap.png umap png格式图片文件
├── 07.CellSplit 步骤7 细胞分割结果目录
│ ├── cell_split_result 细胞分割结果目录
│ │ ├── 0_0.npy 局部细胞分割结果
│ │ ├── 0_0_ori.tif 局部荧光图片
│ │ ├── 0_0.tif 局部细胞识别后荧光图片
… …
│ │ ├── 9500_9500.npy 局部细胞分割结果
│ │ ├── 9500_9500_ori.tif 局部荧光图片
│ │ ├── 9500_9500.tif 局部细胞识别后荧光图片
│ │ ├── all_barcode_num.txt 细胞barcode id对应文件
│ │ ├── all_outline.tif 添加细胞核边界的荧光图片
│ │ ├── cell_color.tif 识别的细胞图片文件
│ │ ├── cellConts.json 识别的细胞json文件
│ │ ├── cells.npy 识别的细胞npy文件
│ │ ├── colors.npy 细胞和颜色对应文件
│ │ ├── conts.tif 细胞分割组织边界信息
│ │ ├── fluorescence.tif 组织荧光图片
│ │ ├── nucleus_color.tif 识别的细胞核图片文件
│ │ ├── nucleusConts.json 识别的细胞核json文件
│ │ ├── nucleus.npy 识别的细胞核pny文件
│ │ ├── progress.txt 进度百分比文件
│ │ └── SegtoBarcode.log 日志文件
│ ├── cluster 聚类结果目录
│ │ ├── cell_cluster_color_img.tif 细胞分割聚类图不含图例tif图片文件
│ │ ├── cell_cluster_color_outline_img.tif 细胞分割聚类图不含图例添加细胞白色边界tif图片文件
│ │ ├── cell_cluster_with_legend_img.png 细胞聚类图含图例png图片文件
│ │ ├── cell_cluster_with_legend_img_small.png 细胞聚类图含图例低分辨率png图片文件
│ │ ├── cell_cluster_with_legend_img.tif 细胞聚类图含图例tif图片文件
│ │ ├── cluster.csv 聚类结果
│ │ ├── cluster_cells_num.csv 聚类类别细胞数统计文件
│ │ ├── clusters_colors.npy 聚类类别和颜色对应结果
│ │ ├── colors.npy 细胞和颜色对应结果
│ │ ├── legend.tif 聚类图例
│ │ ├── marker_gene.csv marker gene信息文件
│ │ ├── object.RDS 细胞分割矩阵得到的Seurat对象结果
│ │ ├── UMAP.pdf umap聚类结果pdf图片文件
│ │ └── UMAP.png umap聚类结果png图片文件
│ ├── images 细胞分割相关图片结果目录
│ │ ├── fluorescence_cell_split.png 荧光图片细胞分割结果png图片文件
│ │ ├── fluorescence_cell_split_small.png 荧光图片细胞分割结果低分辨率png图片文件
│ │ ├── fluorescence_cell_split.tif 荧光图片细胞分割结果tif图片文件
│ │ ├── fluorescence.png 组织荧光png图片文件
│ │ ├── fluorescence_small.png 组织荧光低分辨率png图片文件
│ │ ├── fluorescence.tif 组织荧光tif图片文件
│ │ ├── he_cell_split.png 组织HE染色细胞分割png图片文件
│ │ ├── he_cell_split_small.png 组织HE染色细胞分割低分辨率png图片文件
│ │ ├── he_cell_split.tif 组织HE染色细胞分割tif图片文件
│ │ └── he_hr.tif 组织HE染色tif图片文件
│ └── mtx 细胞分割矩阵结果目录
│ ├── barcodes.tsv.gz 细胞barcode文件
│ ├── cells_center.txt 细胞质心位置文件
│ ├── cells_center.tif 细胞质心图片文件
│ ├── features.tsv.gz 细胞features文件
│ ├── matrix.mtx.gz 细胞矩阵文件
│ └── stat.xls 细胞统计信息文件
├── 08.WebReport 步骤8 网页版报告结果目录
│ ├── src 网页版报告src目录
│ ├── xxx.filelist 生成网页版报告所用的相关文件信息文件
│ ├── xxx.stat.xls 分析结果统计信息文件
│ ├── xxx.rs_stat.xls 分析结果统计信息文件
│ └── xxx.html 网页版报告文件
└── xxx 原始表达矩阵结果目录
├── barcode_pos.tsv barcode及对应芯片位置文件
├── barcode.tsv barcode文件
├── bc_umi_read.tsv.gz barcode对应的umi及reads数文件
├── features.tsv features文件
├── matrix.tsv 矩阵文件
└── umi_gene.tsv.gz barcode对应的umi及基因文件
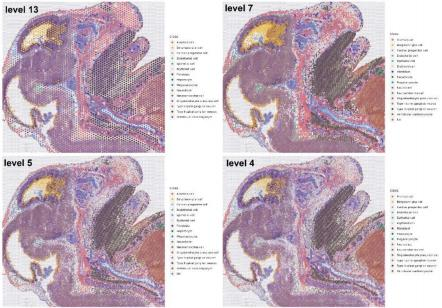
多级分辨率解释
使用 BSTMatrix 分析完数据后,可以得到空间表达矩阵 ,而软件默认会得到 level 1~7 及 level 13 这 8 个 分辨率的结果(05.AllheStat 目录下) 。那么多级分辨率具体是怎么回事呢?下图是多级分辨率实现的说明。level 1 就是单独的 一个 spot 点,level 2 是把中心点及与其相距为(2-1) 的周围 spot 点合并为 一个 ,同理 level 3~13 就是合并周围相应的 level – 1 距离内的 spot 为 一个点。所以单个 spot 的 level 1 对应最高的分辨率水平 5μm,而 level 13 则对应着最低的 100μm。合并后的点称为 supSpot 点。
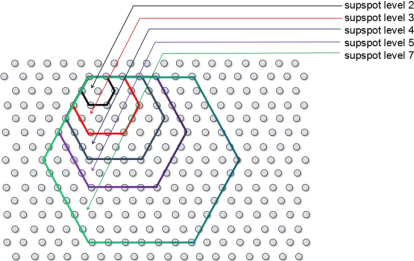
supspot level 与 spot 点阵图
对于空间转录组后续分析并不是使用的分辨率越高越好 。选择合适的分辨率 ,让 spot 点更加接近样本 细胞大小 ,可以使分析结果更接近单细胞分辨率水平 ,还原组织位置上真实的细胞分布 。 空间数据分析分 辨率的选择可以参考以下策略:
A) 冷冻切片等前期实验确定组织中绝大多数细胞的大小 ,后续根据细胞大小选择合适的分辨率;
B) 所选分辨率下 spot点降维聚类可视化结果与切片染色结构最为 一 致的分辨率水平为最佳分辨率;
C) 所选分辨率下 supspot 点的中位基因数达到 1000 以上, 最低不低于 500。
例如, 下图中的 S1000 空间转录组可以选择 L2( 10μm)作为合适的分辨率水平进行后续分析。
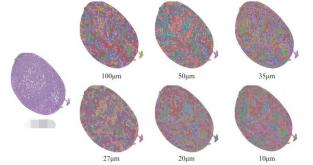
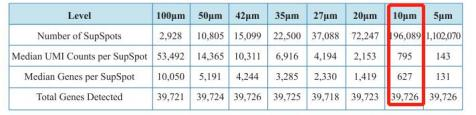
后续分析
3.1 seurat 对象创建
百创 S1000 空间转录组矩阵图像信息与 10X 空间矩阵不同, 不方便直接使用 Seurat 空间作图函数进行作 图,根据 S1000 空间矩阵创建 10x 空间 seurat 对象格式,使用该方法创建 object 对象,可使用 SpatialFeaturePlot 等 seurat 内置空间作图函数来进行分析
脚本:
CreateBmkObject.R
用法:
source(“CreateBmkObject.R”)
object <- CreateS1000Object(
matrix_path=”BSTViewer_project/subdata/L13_heAuto”, #矩阵文件目录
png_path=”BSTViewer_project/he_roi_small.png”, #png 格式图片路径
spot_radius =1, #点的半径 ,可以不指定 ,会自动计算
min.cells = 5, # 一个基因至少在 n 个细胞中表达才被保留 ,可自行调整 ,默认值 5
min.features = 100 # 一个细胞至少有 n 个基因才被保留 ,可自行调整 ,默认值 100 )
【脚本见附件】
3.2 人/ 小鼠 SingleR 自动注释
脚本: /share/nas1/lixiaolei/bin/SingleR.r
参数:
| -i INDIR, –indir=INDIR | 矩阵所在目录 |
| -o OUTDIR, –outdir=OUTDIR | 输出目录 |
| –he_png=HE_PNG | png 格式 he 文件 |
| –res=RES | resolution, default 0.5 |
| –MinCell=MINCELL | MinCell, default 5 |
| –MinFeatures=MINFEATURES | MinFeatures, default 100 |
| –point_size=POINT_SIZE | point_size, default 1 |
| –ref=REF | SingleR 自 带 参 考 数 据 集 , 1 for BlueprintEncodeData;2 for |
DatabaseImmuneCellExpressionData;3 for NovershternHematopoieticData;4 for MonacoImmuneData;5 for HumanPrimaryCellAtlasData;6 for ImmGenData;7 for MouseRNAseqData,default 1
-h, –help Show this help message and exit
输出:
singleR/
├── cluster.csv
├── singleR_cluster.pdf
├── singleR_cluster.png
├── singleR_cluster_umap.p* ├── singleR_UMAP.p*
#聚类结果以及 SingleR 注释信息
#SingleR 注释结果空间位置图(pdf 格式)
#SingleR 注释结果空间位置图(png 格式)
#SingleR 空间位置图+umap 图(pdf/png 格式)
#SingleR umap 图(pdf/png 格式)
└── UMAP.p* #聚类结果 umap 图(pdf/png 格式)
【脚本见附件】
3.3 百创 S1000 空间转录组不同细胞类型相邻边界 barcode 提取
组织内部亚群并不独立,在彼此接触的范围可能会发生广泛的配体- 受体相互作,对空间转录组进行细 胞类型注释后,可以提取不同细胞类型相邻边界的 barcode 进行细胞通讯或互作分析,以下脚本为如何提取 不同细胞类型相邻边界的 barcode。
脚本:bianjie_script.r
参数:
-i 参数指定输入目录 ,例:BSTViewer_project/subdata/L13_heAuto
-o 参数指定输出目录
–he_png 参数指定图像路径 ,例:BSTViewer_project/he_roi_small.png
–point_size 参数指定作图点的大小
–cluster 参数指定细胞注释文件, 第 一 列为 barcode 信息, 第 二 列为细胞注释信息 ,表头为 Barcode, singleR
–cell_type1 参数指定第 一种细胞类型
–cell_type2 参数指定第二种细胞类型
输出 :指定细胞相邻边界 barcode 空间位置图片

【脚本见附件】
3.4 细胞通讯分析
细胞通讯分析的基础是基因表达数据和配体- 受体数据库信息 ,例如转录组数据表明 A、B 细胞分别表 达了基因α和β , 通过数据库查询α和β是配体- 受体关系, 则认为 A-B 通过α- β途径进行了通讯。
3.4.1 输入数据格式要求
A) 细胞注释文件(meta.txt,两列,分别为细胞 id 和细胞类型)
B) 基因表达矩阵(txt)
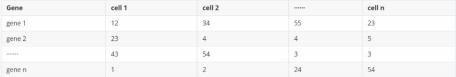
3.4.2 软件安装
# 下载软件(按顺序安装)
conda create -n cellphonedb2 python=3.7
conda install Cython
conda install h5py
conda install scikit-learn
conda install r
conda install rpy2
pip install https://pypi.tuna.tsinghua.edu.cn/simple cellphonedb
pip install -i https://pypi.tuna.tsinghua.edu.cn/simple markupsafe==2.0.1
#R 包
install.packages(‘ggplot2’)
install.packages(‘heatmap’)
#data1—> R
#使用 RCTD 注释的空间数据进行后续分析
results <- myRCTD@results
results_df <- results$results_df
barcodes = rownames(results_df[results_df$spot_class != “reject” & puck@nUMI >= 1,])
my_table = puck@coords[barcodes,]
my_table$class = results_df[barcodes,]$first_type
meta = my_table %>% select(class) %>% rownames to column(var = ‘cell’) %>% dplyr::rename(cell_type = class) write.table(meta ,file = ‘/xxx/Test/meta.txt’, sep = ‘\t’, quote = F, row.names = T)
#data2—> R
expr <- Read10X(‘/xxx/BSTViewer_project/subdata/L13_heAuto/’,cell.column = 1)
object <- CreateSeuratObject(counts = expr,assay = “Spatial”)
counts = object[[‘Spatial’]]@counts %>% as.data.frame() %>% rownames to column(var = ‘Gene’) write.table(counts ,file = ‘/xxx/Test/counts.txt’, sep = ‘\t’, quote = F, row.names = T)
#鼠源基因转化
#CellPhoneDB 只能识别人类基因 ,如果是鼠源基因需要进行同源基因的转化
# 下载人鼠同源基因对应关系
http://asia.ensembl.org/biomart/martview/b9f8cc0248e4714ba8e0484f0cbe4f02
#参考步骤
https://www.jianshu.com/p/922a7f2c21fc
# 下载的对应关系表应该有四列
Gene_stable_ID | Gene_name | Mouse_gene_stable_ID | Mouse_gene_name
#将对应关系(human_mouse_gene.txt)替换到 data2 矩阵—>Shell
awk -F ‘\t’ ‘BEGIN{OFS == ‘\t’}ARGIND==1 {a[$4]=$1}ARGIND==2 {if(FNR ==1){print}else {$1 = a[$1];print} }’ human_mouse_gene.txt counts.txt | sed ‘/^ /d’ | sed ‘1s/^/Gene\t/’ | sed ‘s/\s/\t/g’ > counts_new.txt
#从远程存储库中列出 cellphonedb 可用数据库版本:
cellphonedb database list_remote
#查看本地数据库
cellphonedb database list_local
# 下载远程数据库
cellphonedb database download –version <version_spec |latest>
#运行 cellphonedb —> Shell
cellphonedb method statistical_analysis –output-path test_output meta.txt counts_new.txt
#输出文件
deconvoluted.txt | pvalues.txt | significant_means.txt
test.count_network.txt | means.txt | test.interaction_count.txt
#气泡图—> Shell
cellphonedb plot dot_plot –pvalues-path test_output/pvalues.txt –means-path test_output/means.txt –output-path te st_output –output-name test.dotplot.pdf
#仅绘制自己关注的细胞和互作—> Shell
cellphonedb plot dot_plot –pvalues-path test_output/pvalues.txt –means-path test_output/means.txt –output-path te st_output –output-name test.dotplot2.pdf–rows test_output/row.txt –columns test_output/col.txt

注 :横坐标是细胞类型互作 ,纵坐标是蛋白互作 ,点越大表示 p 值越小,颜色代表平均表达量
#热图—> Shell
cellphonedb plot heatmap_plot –pvalues-path test_output/pvalues.txt –output-path test_output –pvalue 0.05 –count -name test.heatmap_count.pdf –log-name test.heatmap_log_count.pdf –count-network-name test.count_network.txt – -interaction-count-name test.interaction_count.txt meta.txt

注:横纵坐标表示细胞类型,颜色深蓝色到紫红色代表互作数由低到高
# 细胞类型网络图—> R
#这里借用另一款细胞通讯分析软件——CellChat(https://github.com/sqjin/CellChat)的绘图方法 #加载 R 包
library(igraph)
library(tidyr)
library(stats)
library(reshape2)
library(Matrix)
library(grDevices)
library(ggplot2)
#使用 CellChat 作者提供的绘制互作网络图的 function, 复制粘贴即可
scPalette <- function(n) {
colorSpace <- c(‘#E41A1C’,’#377EB8′,’#4DAF4A’,’#984EA3′,’#F29403′,’#F781BF’,’#BC9DCC’,’#A65628′,’#54B0 E4′,’#222F75′,’#1B9E77′,’#B2DF8A’,
‘#E3BE00′,’#FB9A99′,’#E7298A’,’#910241′,’#00CDD1′,’#A6CEE3′,’#CE1261′,’#5E4FA2′,’#8CA77B’,’# 00441B’,’#DEDC00′,’#B3DE69′,’#8DD3C7′,’#999999′)
if (n <= length(colorSpace)) {
colors <- colorSpace[1:n]
} else {
colors <- grDevices::colorRampPalette(colorSpace)(n)
}
return(colors)
}
netVisual_circle <-function(net, color.use = NULL,title.name = NULL, sources.use = NULL, targets.use = NULL, id ents.use = NULL, remove.isolate = FALSE, top = 1,
weight.scale = FALSE, vertex.weight = 20, vertex.weight.max = NULL, vertex.size.max = NULL, v ertex.label.cex=1,vertex.label.color= “black”,
edge.weight.max = NULL, edge.width.max=8, alpha.edge = 0.6, label.edge = FALSE,edge.label.colo r=’black’,edge.label.cex=0.8,
edge.curved=0.2,shape=’circle’,layout=in_circle(), margin=0.2, vertex.size = NULL,
arrow.width=1,arrow.size = 0.2){
if (!is.null(vertex.size)) {
warning(“‘vertex.size’ is deprecated. Use `vertex.weight`”)
}
if (is.null(vertex.size.max)) {
if (length(unique(vertex.weight)) == 1) {
vertex.size.max <- 5
} else {
vertex.size.max <- 15
}
}
options(warn = -1)
thresh <- stats::quantile(net, probs = 1-top)
net[net < thresh] <- 0
if ((!is.null(sources.use)) | (!is.null(targets.use)) | (!is.null(idents.use)) ) {
if (is.null(rownames(net))) {
stop(“The input weighted matrix should have rownames!”)
}
cells.level <- rownames(net)
df.net <- reshape2::melt(net, value.name = “value”)
colnames(df.net)[1:2] <- c(“source”,”target”)
# keep the interactions associated with sources and targets of interest
if (!is.null(sources.use)){
if (is.numeric(sources.use)) {
sources.use <- cells.level[sources.use]
}
df.net <- subset(df.net, source %in% sources.use)
}
if (!is.null(targets.use)){
if (is.numeric(targets.use)) {
targets.use <- cells.level[targets.use]
}
df.net <- subset(df.net, target %in% targets.use)
}
if (!is.null(idents.use)) {
if (is.numeric(idents.use)) {
idents.use <- cells.level[idents.use]
}
df.net <- filter(df.net, (source %in% idents.use) | (target %in% idents.use))
}
df.net$source <- factor(df.net$source, levels = cells.level)
df.net$target <- factor(df.net$target, levels = cells.level)
df.net$value[is.na(df.net$value)] <- 0
net <- tapply(df.net[[“value”]], list(df.net[[“source”]], df.net[[“target”]]), sum)
}
net[is.na(net)] <- 0
if (remove.isolate) {
idx1 <- which(Matrix::rowSums(net) == 0)
idx2 <- which(Matrix::colSums(net) == 0)
idx <- intersect(idx1, idx2)
net <- net[-idx, ]
net <- net[, -idx]
}
g <- graph_from_adjacency_matrix(net, mode = “directed”, weighted = T)
edge.start <- igraph::ends(g, es=igraph::E(g), names=FALSE)
coords<-layout_(g,layout)
if(nrow(coords)!=1){
coords_scale=scale(coords)
}else {
coords_scale<-coords
}
if (is.null(color.use)) {
color.use = scPalette(length(igraph::V(g)))
}
if (is.null(vertex.weight.max)) {
vertex.weight.max <- max(vertex.weight)
}
vertex.weight <- vertex.weight/vertex.weight.max*vertex.size.max+5
loop.angle<-ifelse(coords_scale [igraph::V(g),1]>0,-atan(coords_scale [igraph::V(g),2]/coords_scale [igraph::V(g),1]),pi-a tan(coords_scale [igraph::V(g),2]/coords_scale [igraph::V(g),1]))
igraph::V(g)$size<-vertex.weight
igraph::V(g)$color<-color.use[igraph::V(g)]
igraph::V(g)$frame.color <- color.use[igraph::V(g)]
igraph::V(g)$label.color <- vertex.label.color
igraph::V(g)$label.cex<-vertex.label.cex
if(label.edge){
igraph::E(g)$label<-igraph::E(g)$weight
igraph::E(g)$label <- round(igraph::E(g)$label, digits = 1)
}
if (is.null(edge.weight.max)) {
edge.weight.max <- max(igraph::E(g)$weight)
}
if (weight.scale == TRUE) {
#E(g)$width<-0.3+edge.width.max/(max(E(g)$weight)-min(E(g)$weight))*(E(g)$weight-min(E(g)$weight)) igraph::E(g)$width<- 0.3+igraph::E(g)$weight/edge.weight.max*edge.width.max
}else {
igraph::E(g)$width<-0.3+edge.width.max*igraph::E(g)$weight
}
igraph::E(g)$arrow.width<-arrow.width
igraph::E(g)$arrow.size<-arrow.size
igraph::E(g)$label.color<-edge.label.color
igraph::E(g)$label.cex<-edge.label.cex
igraph::E(g)$color<- grDevices::adjustcolor(igraph::V(g)$color[edge.start[,1]],alpha.edge)
if(sum(edge.start[,2]==edge.start[,1])!=0){
igraph::E(g)$loop.angle [which(edge.start[,2]==edge.start[,1])]<-loop.angle [edge.start[which(edge.start[,2]==edge.star t[,1]),1]]
}
radian.rescale <- function(x, start=0, direction=1) {
c.rotate <- function(x) (x + start) %% (2 * pi) * direction
c.rotate(scales::rescale(x, c(0, 2 * pi), range(x)))
}
label.locs <- radian.rescale(x=1:length(igraph::V(g)), direction=-1, start=0)
label.dist <- vertex.weight/max(vertex.weight)+2
plot(g,edge.curved=edge.curved,vertex.shape=shape,layout=coords_scale,margin=margin, vertex.label.dist=label.dist, vertex.label.degree=label.locs, vertex.label.family=”Helvetica”, edge.label.family=”Helvetica”) # “sans”
if (!is.null(title.name)) {
text(0,1.5,title.name, cex = 1.1)
}
# https://www.andrewheiss.com/blog/2016/12/08/save-base-graphics-as-pseudo-objects-in-r/ # grid.echo()
# gg <- grid.grab()
gg <- recordPlot()
return(gg)
}
#整体互作网络图绘制
#读取 cellphonedb 的分析数据
count_net <- read.delim(“/xxx/test_output/test.count_network.txt”, check.names = FALSE)
#数据格式处理
count_inter <- count_net
count_inter$count <- count_inter$count/100
count_inter <- spread(count_inter, TARGET, count)
rownames(count_inter) <- count_inter$SOURCE
count_inter <- count_inter[, -1]
count_inter <- as.matrix(count_inter)
#绘图
netVisual_circle(count_inter,weight.scale = T)
#每种细胞与其他细胞的互作
par(mfrow = c(1,3), xpd=TRUE)
for (i in 1:nrow(count_inter)) {
mat2 <- matrix(0, nrow = nrow(count_inter), ncol = ncol(count_inter), dimnames = dimnames(count_inter))
mat2[i, ] <- count_inter[i, ]
netVisual_circle(mat2,
weight.scale = T,
edge.weight.max = max(count_inter),
title.name = rownames(count_inter)[i],
arrow.size=0.2)
}
3.5 单细胞空间联合分析
软件 RCTD
使用监督学习的方法 ,利用带注释的 scRNA-seq 数据来定义空间转录组学数据中预期存在的细胞类型 的细胞特异性情况。
3.5.1 单细胞数据格式要求
A)data 1 细胞注释结果( cellType)
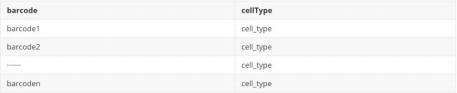
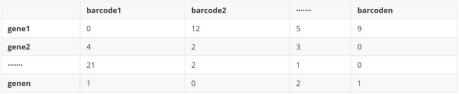
B)data2 基因表达矩阵( sc_counts)
3.5.2 空间数据格式要求
A) data1空间 spots 位置信息( coords)
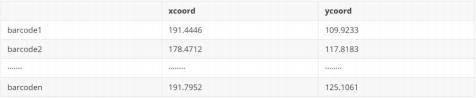
B)data2 基因表达矩阵( sp_counts)
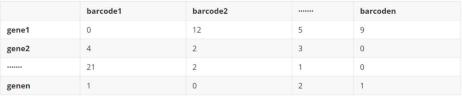
#安装 spacexr 包
# install.packages(“devtools”)
devtools::install_github(“dmcable/spacexr”, build_vignettes = FALSE)
#加载 R 包
library(Seurat)
library(tidyverse)
library(Matrix)
library(spacexr)
library(ggplot2)
library(ggpubr)
library(gridExtra)
library(reshape2)
library(readr)
library(Seurat)
library(config)
library(ggpubr)
library(gridExtra)
library(reshape2)
library(png)
library(patchwork)
library(SingleR)
library(celldex)
#矩阵转化
Rcpp::sourceCpp(code=’
#include <Rcpp.h>
using namespace Rcpp;
// [[Rcpp::export]]
IntegerMatrix asMatrix(NumericVector rp,
NumericVector cp,
NumericVector z,
int nrows,
int ncols){
int k = z.size() ;
IntegerMatrix mat(nrows, ncols);
for (int i = 0; i < k; i++){
mat(rp[i],cp[i]) = z [i];
}
return mat;
}
‘ )
as_matrix <- function(mat){
row_pos <- mat@i
col_pos <- findInterval(seq(mat@x)-1,mat@p[-1])
tmp <- asMatrix(rp = row_pos, cp = col_pos, z = mat@x,
nrows = mat@Dim[1], ncols = mat@Dim[2])
row.names(tmp) <- mat@Dimnames[[1]]
colnames(tmp) <- mat@Dimnames[[2]]
return(tmp)
}
#读取单细胞数据
expr <- Read10X(‘/xxx/04.QC/filtered_feature_bc_matrix/’, cell.column = 1)
sc_data <- CreateSeuratObject(counts = expr,assay = “RNA”,min.cells=5,min.features=100)
#counts & nUMI
sc_counts <- as_matrix(sc_data[[‘RNA’]]@counts)
sc_nUMI = colSums(sc_counts)
# 注释
##方法 一:SingleR 注释
load(‘/share/nas1/guochao/database/celldex/MouseRNAseqData.Rdata’)
mouseRNA <- ref
sce_for_SingleR<-GetAssayData(sc_data,slot=”data”)
pred.mouseRNA <- SingleR(test=sce_for_SingleR,ref=mouseRNA,labels=mouseRNA$label.fine,assay.type.test=”logc ounts”,assay.type.ref =”logcounts”)
pred.mouseRNA$labels<-as.factor(pred.mouseRNA$labels)
cellType=data.frame(barcode=sc_data@assays$RNA@counts@Dimnames[2], celltype=pred.mouseRNA$labels) names(cellType) = c(‘barcode’, ‘cell_type’)
cell_types = cellType$cell_type; names(cell_types) <- cellType$barcode # create cell_types named list cell_types = as.factor(cell_types) # convert to factor data type
##方法二:手动注释
sc_data = NormalizeData(sc_data, normalization.method = ‘LogNormalize’, scale.factor = 10000) sc_data = FindVariableFeatures(sc_data, selection.method = ‘vst’, nfeatures = 2000)
sc_data = ScaleData(sc_data)
sc_data = RunPCA(sc_data, features = VariableFeatures(object = sc_data))
sc_data = FindNeighbors(sc_data, dims = 1:15)
sc_data = FindClusters(sc_data, resolution = 0.25)
markers = FindAllMarkers(sc_data, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
top10 = markers %>% group_by(cluster) %>% top_n(n = 5, wt = avg_log2FC)
my_df = sc_data@meta.data %>% as.data.frame() %>%
select(seurat_clusters) %>%
rownames to column(var = ‘barcode’) %>%
rename(cluster = seurat_clusters)
cellType = my_df %>% mutate(cell_type = case_when( cluster == ‘0’ ~ ‘Allantois cell’,
cluster == ‘1’ ~ ‘Bergmann glial cell’,
cluster == ‘2’ ~ ‘Neuroblast’,
cluster == ‘3’ ~ ‘Neuroendocrine cell’,
cluster == ‘4’ ~ ‘Fibroblast’,
cluster == ‘5’ ~ ‘Type I spiral ganglion neuron’,
cluster == ‘6’ ~ ‘Erythroid cell’,
cluster == ‘7’ ~ ‘Type II spiral ganglion neuron’,
cluster == ‘8’ ~ ‘Epithelial cell’,
cluster == ‘9’ ~ ‘Cardiac progenitor cell’,
cluster == ’10’ ~ ‘Oligodendrocyte precursor cell’,
cluster == ’11’ ~ ‘Endothelial cell’,
cluster == ’12’ ~ ‘Megakaryocyte’,
cluster == ’13’ ~ ‘Hepatocyte’,
cluster == ’14’ ~ ‘Ventricular cardiomyocyte’))
cell_types = cellType$cell_type; names(cell_types) <- cellType$barcode # create cell_types named list cell_types = as.factor(cell_types) # convert to factor data type
#构建参考集
reference = Reference(sc_counts, cell_types, sc_nUMI)
#读取空间数据 ,这里选择的是level 13 的数据
#位置信息
coords = read.table(gzfile(‘/xxx/05.AllheStat/BSTViewer_project/subdata/L13_heAuto/barcodes_pos.tsv.gz’), hea der = F) %>% dplyr::rename(barcodes = V1, xcoord = V2, ycoord = V3)
rownames(coords) <- coords$barcodes; coords$barcodes <- NULL
#表达量矩阵
expr <- Read10X(‘/xxx/05.AllheStat/BSTViewer_project/subdata/L13_heAuto/’, cell.column = 1) sp_data <- CreateSeuratObject(counts = expr,assay = “Spatial”)
sp_counts <- as_matrix(sp_data[[‘Spatial’]]@counts)
#nUMI
sp_nUMI <- colSums(sp_counts)
#构建空间实验集
puck <- SpatialRNA(coords, sp_counts, sp_nUMI)
#联合分析
myRCTD <- create.RCTD(puck, reference, max_cores = 8)
myRCTD <- run.RCTD(myRCTD, doublet_mode = ‘doublet’) #run.RCTD
#查找 marker 基因
get_marker_data <- function(cell_type_names, cell_type_means, gene_list) {
marker_means = cell_type_means[gene_list,]
marker_norm = marker_means / rowSums(marker_means)
marker_data = as.data.frame(cell_type_names[max.col(marker_means)])
marker_data$max_epr <- apply(cell_type_means[gene_list,],1,max)
colnames(marker_data) = c(“cell_type”,’max_epr’)
rownames(marker_data) = gene_list
marker_data$log_fc <- 0
epsilon <- 1e-9
for(cell_type in unique(marker_data$cell_type)) {
cur_genes <- gene_list[marker_data$cell_type == cell_type]
other_mean = rowMeans(cell_type_means[cur_genes,cell_type_names != cell_type])
marker_data$log_fc [marker_data$cell_type == cell_type] <- log(epsilon + cell_type_means[cur_genes,cell_type]) – l og(epsilon + other_mean)
}
return(marker_data)
}
cell_type_info_restr = myRCTD@cell_type_info$info
de_genes <- get_de_genes(cell_type_info_restr, puck, fc_thresh = 3, expr_thresh = .0001, MIN_OBS = 3) marker_data_de = get_marker_data(cell_type_info_restr[[2]], cell_type_info_restr[[1]], de_genes)
saveRDS(marker_data_de,’/share/nas1/guochao/Test/221107marker_data_de_standard.RDS’)
write.table(marker_data_de, file = ‘/share/nas1/guochao/Test/221107marker_data_de_standard.tsv’, sep = ‘\t’,quote = F, row.names = T)
#构建绘图数据
results <- myRCTD@results
results_df <- results$results_df
barcodes = rownames(results_df[results_df$spot_class != “reject” & puck@nUMI >= 1,])
my_table = puck@coords[barcodes,]
my_table$class = results_df[barcodes,]$first_type
#绘图
cal_zoom_rate <- function(width, height){
std_width = 1000
std_height = std_width / (46 * 31) * (46 * 36 * sqrt(3) / 2.0)
if (std_width / std_height > width / height){
scale = width / std_width
}
else {
scale = height / std_height
}
return(scale)
}
png <- readPNG(‘/share/nas1/dengdj/testing/Barcode_YF/20220923-YF-N1295-1-2/analysis/XSPT-T/05.AllheSta t/allhe/he_roi_small.png’)
zoom_scale = cal_zoom_rate(dim(png)[2], dim(png)[1])
my_table = my_table %>% mutate(across(c(x,y), ~.x*zoom_scale))
col = c(“#F56867″,”#FEB915″,”#C798EE”,”#59BE86″,”#7495D3″,”#D1D1D1″,”#6D1A9C”,”#15821E”,”#3A84 E6″,”#997273″,”#787878″,”#DB4C6C”,”#9E7A7A”,”#554236″,”#AF5F3C”,”#93796C”,”#F9BD3F”,”#DAB370″)
p = ggplot(my_table, aes(x = x, y = dim(png)[1] – y))+
background_image(png)+
geom_point(shape = 16, size = 1.8, aes(color = class))+
coord_cartesian(xlim = c(0, dim(png)[2]), y = c(0, dim(png)[1]), expand = FALSE)+
scale_color_manual(values = col)+
theme(axis.title.x=element_blank(),axis.text.x=element_blank(),axis.ticks.x=element_blank(), axis.title.y=element_bl ank(),axis.text.y=element_blank(),axis.ticks.y=element_blank())+
guides(color = guide_legend(override.aes = list(size = 2.5, alphe = 0.1)))
ggsave(p, file = ‘/share/nas1/guochao/Test/temp/221107_all.png’, width=10, height=7, dpi = 300) ggsave(p, file = ‘/share/nas1/guochao/Test/temp/221107_all.pdf’, width=10, height=7)

{kind=link}
{kind=link}
{kind=link}
{kind=link}